Visit these blogs to read about families who have adopted a child with OI:
Journey to Olivia
The O’Cain Crew
Adopting a Child with Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a genetic disorder characterized by fragile bones that break easily. Also known as “brittle bone disease,” a person is born with this disorder and is affected through his or her lifetime. As a parent of a child/children with OI, our job is to encourage, empower, and equip them, and not succumb to the temptation to cover them in bubble wrap.
In addition to fractures people with OI may have muscle weakness, hearing loss, fatigue, joint laxity (loose and very flexible joints), curved bones, scoliosis, blue sclera (the whites of your eyes), dentinogenesis imperfecta (DI/brittle teeth), and short stature. Restrictive pulmonary disease occurs in more severely affected people.
OI is caused by an “error” or a mutation on a gene that affects the body’s production of the collagen found in bones, and other tissues. It is not caused by too little calcium or poor nutrition. Collagen act as the shock absorbers in your bones. Because the collagen is faulty, when the bones are bumped, there is nothing to cushion them, and so they break. Approximately 35% of children with OI are born into a family with no family history of OI. Most often this is due to a new mutation to a gene and not by anything the parents did before or during pregnancy.
OI is variable with 8 different types. The types range in severity from a lethal form to a milder form with few visible symptoms. The specific medical problems a person will encounter will depend on the degree of severity. A person with mild OI may experience a few fractures (there are some OI kids who play lacrosse!) while those with the severe forms may have hundreds in a lifetime (and spend their time in a wheelchair). The type of OI an adopted child has will probably not be known. Knowing the severity (mild, moderate or severe) of the child’s OI is helpful but may not be accurate if stated from the country of birth.
Diagnosis for OI is primarily based on signs seen in a doctor’s examination. When there is uncertainty about the diagnosis, it is best to consult a physician who is familiar with OI. Genetic testing is available to confirm a diagnosis of OI through collagen or gene analysis—a skin sample or a blood sample are used. While this is a nice thing to know, it is expensive and not necessary.
The OI type descriptions provide general information about how severe the symptoms probably will be. Health issues frequently seen in children and adults who have OI include:
• Short stature
• Weak tissues, fragile skin, muscle weakness, and loose joints
• Bleeding, easy bruising, frequent nosebleeds and in a small number of people heavy bleeding from injuries
• Hearing loss may begin in childhood and affects approximately 50% of adults
• Breathing problems, higher incidence of asthma plus risk for other lung problems
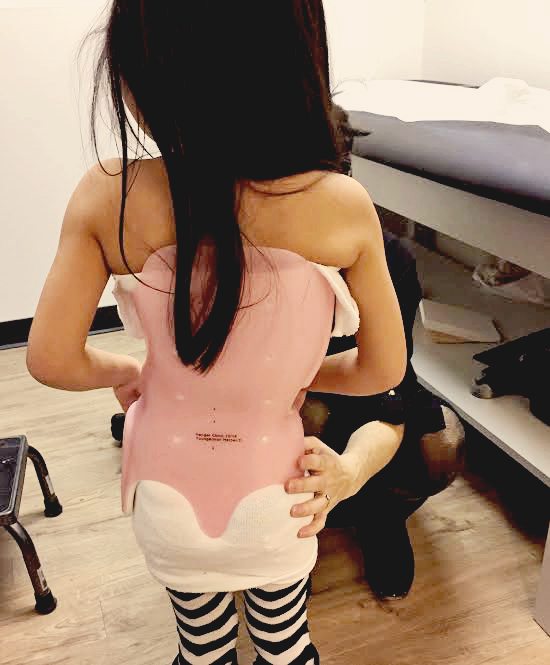
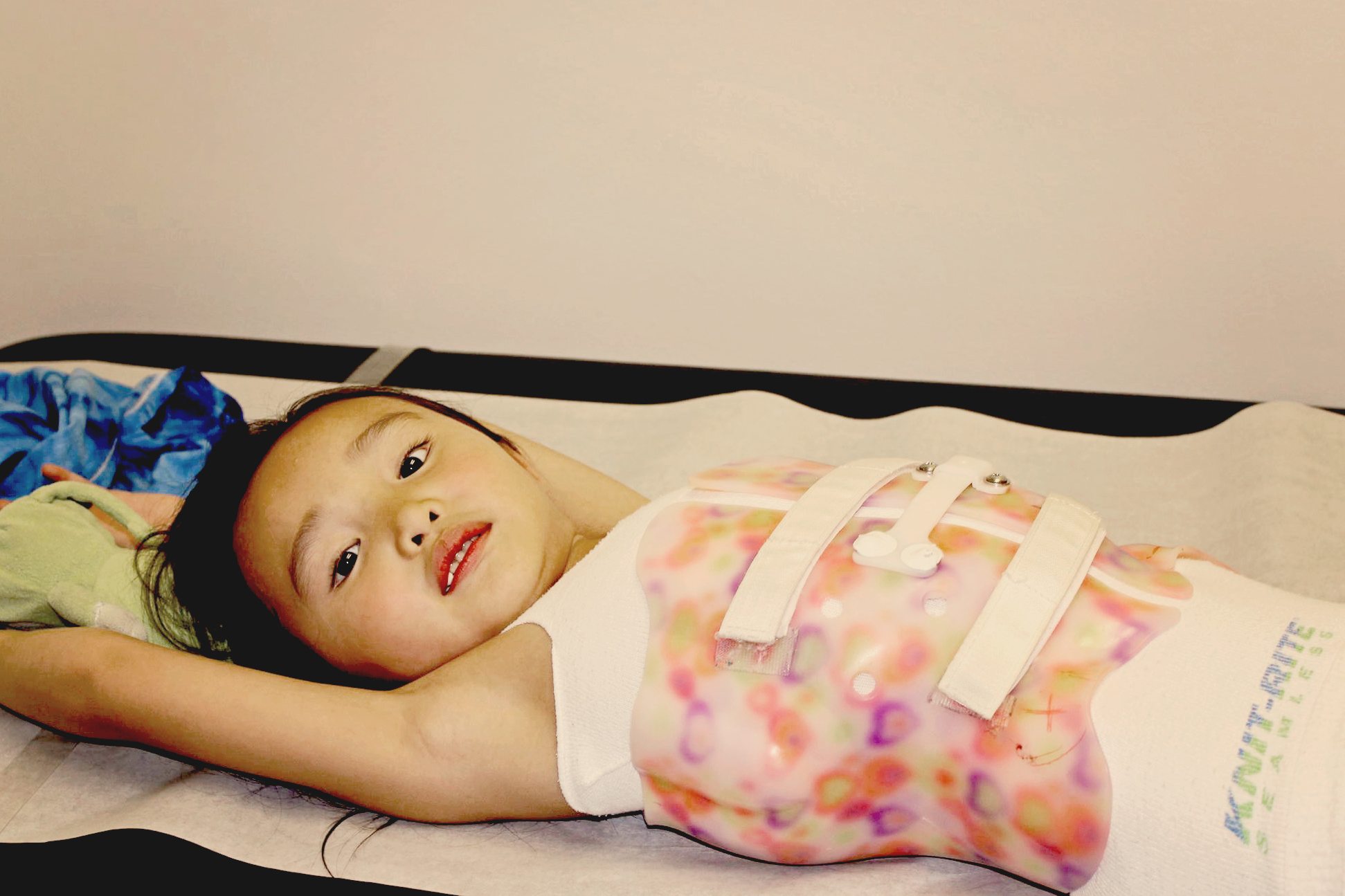
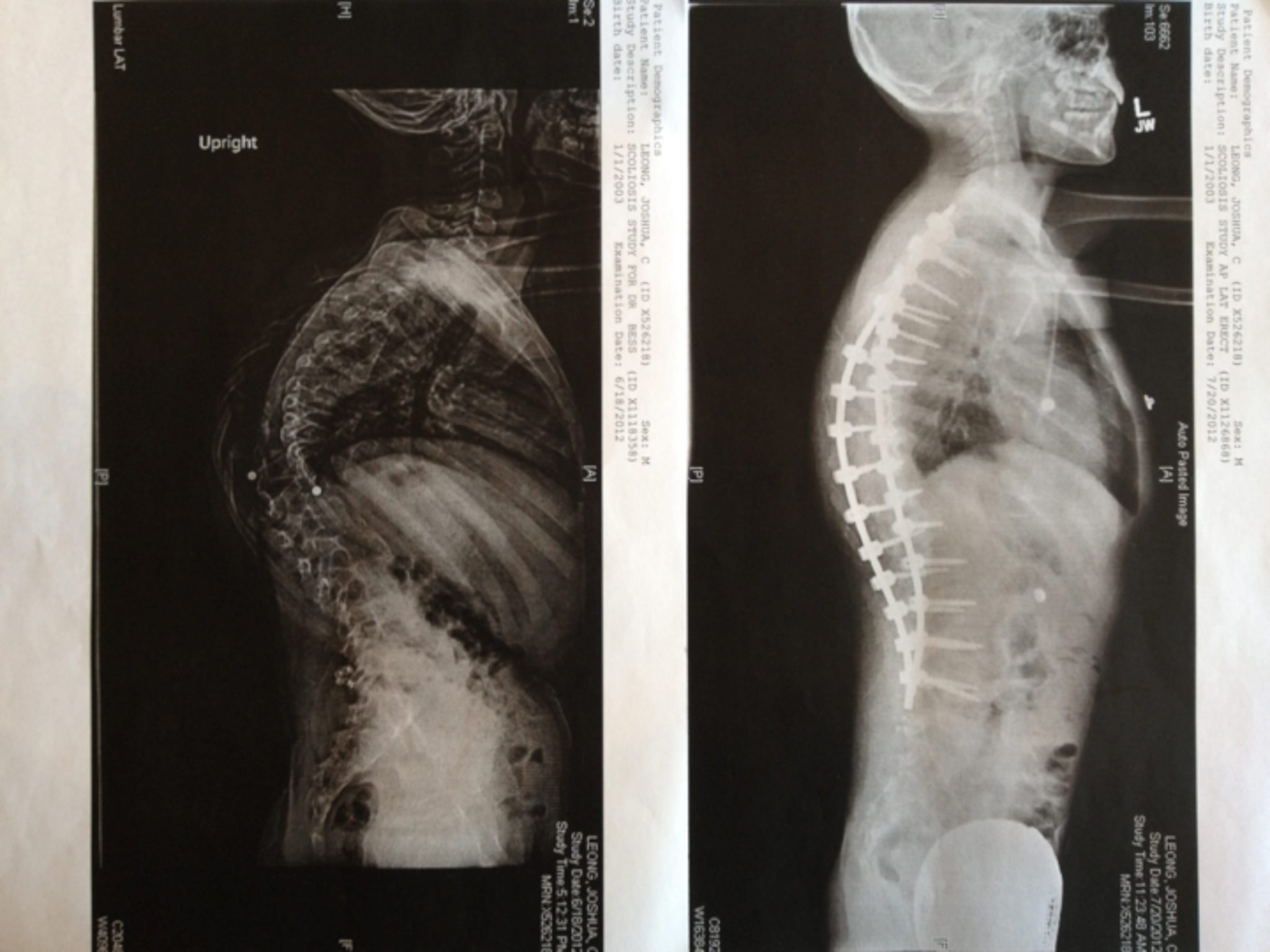
• Curvature of the spine
Doctors who see children and adults with OI include primary care physicians, orthopedists, endocrinologists, geneticists and physiatrists (rehabilitation specialists). Other specialists such as a neurologist may be needed. Most Shriners hospitals have seen OI patients. Of course the more they see the better informed they are in treating. In my (Jewel’s) experience they also do their best to assist you in being as independent as you are comfortable. In other words, if you can splint or wrap at home and don’t need to be seen except for extreme breaks, they can help you deal with that.
Treatments focuses on minimizing fractures, maximizing mobility, maximizing independent function and general health; physical therapy and safe exercise including swimming; casts, splints or wraps for broken bones; braces to support legs, ankles, knees and wrists as needed; orthopedic surgery, often including implanting rods to support the long bones in arms or legs; medications to strengthen bones; and mobility aids such as canes, walkers, or wheelchairs and other equipment or aids for independence may be needed to compensate for weakness or short stature.
At this time, there is no cure. The prognosis for a person with OI varies greatly depending on the number and severity of symptoms. Life expectancy is not affected in people with mild or moderate symptoms, it may however, be shortened for those with more severe symptoms. The most severe forms result in death at birth or during infancy. Respiratory failure is the most frequent cause of death for people with OI, followed by accidental trauma. Despite the challenges of managing OI, most adults and children who have OI lead productive and successful lives. They attend school, develop friendships and other relationships, have careers, raise families, participate in sports (track, see below) and other recreational activities and are active members of their communities (Source).
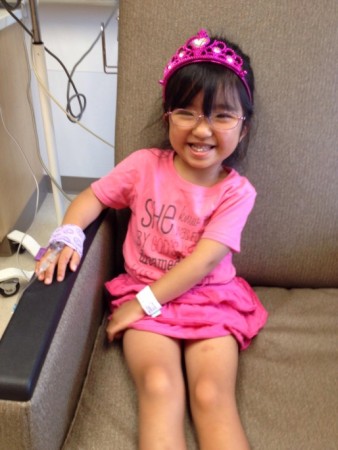
Note from Jewel, a mother who has adopted a daughter with OI (pictured below) –
“Personally, my daughter with OI is a joy to be around! She was adopted at 8 years old and is now 9. She expresses sorrow for her friend (who has yet to be adopted) who is afraid to stand on his own and perhaps walk. She understands the fear but she says, ‘I would try to walk!’ And she is trying now that her legs have rods and are straight, it won’t be long now. While she will probably never walk without some assistance, cane or walker, she will have the freedom to stand and walk as tolerated. She told me once that she would rather hurt herself and experience things than to just sit at home and do nothing except be “safe.” A family provides a stable, consistent environment of care for an adopted child with OI. If they did receive treatment, drugs or surgeries in their birth country, that treatment was probably not given as consistently as mom or dad will work to provide and would not be adequate care!”
Note from Jen, a mother who has adopted a daughter with OI (pictured above):
“My daughter was 4.5 when I adopted her from China. I knew that she was relatively mild, but did not know the type. Her left femur was bowed enough that there was 1.5” difference between her 2 legs. On our 1 month home anniversary, her heel bumped at just the right angle and it broke. But it was a blessing. Our orthopedic surgeon was able to rod it and his expertise is bone lengthening. The leg is now completely straight and we gained back the lost leg length difference. Olivia also has DI and has had caps put on to protect some of her teeth. She lost her first tooth, and has 2 adult teeth coming in. She has responded very well to her infusions that she receives every 3 months (we receive zolandronic acid, the other drug option is pamidronate). Olivia has grown in height, grown in strength, runs, jumps, and loves to dance. She is one of the most positive and compassionate people I’ve ever met (not to mention most competitive). Give her a challenge and she will rise to the occasion!”

Resources:
OI Foundation
Nationwide Children’s Hospital: What is OI? and The Metabolic Bone Clinic
Shriners Hospitals
Kennedy Krieger Institute
Brittle Bone Society
All I Need- Osteogenesis Imperfecta
OI Parents Facebook Group
Parents of Adopted Children with OI Facebook Group
Read blog posts about Osteogenesis Imperfecta (OI) on No Hands But Ours.