Visit these blogs to read about families who have adopted children with this special need:
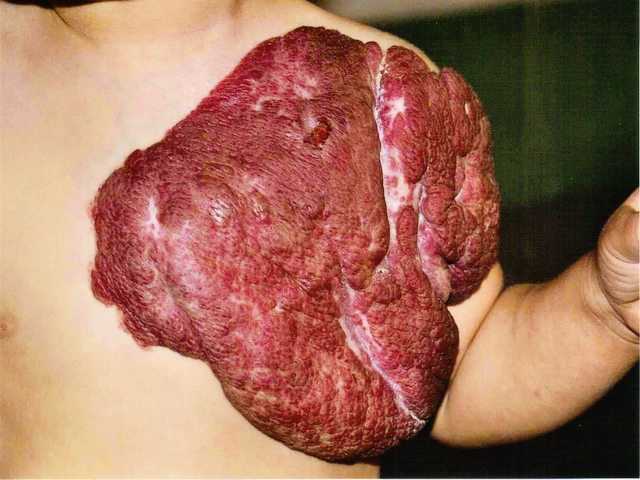
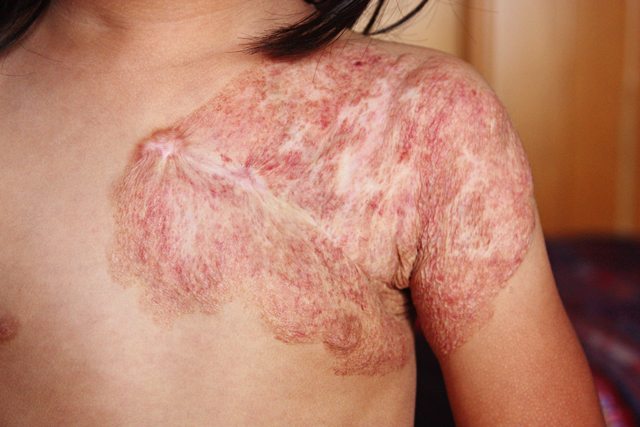
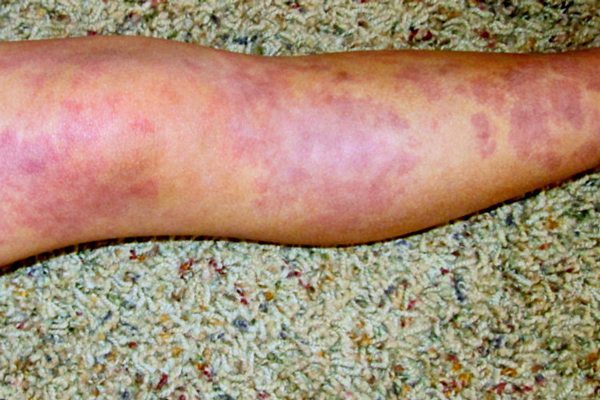
Capillary malformations, commonly known as port wine stains, occur in about 0.3% of live births. These flat, well defined areas range from pale pink to dark purple and are present at birth. Port wine stains may occur anywhere on the body, but they are predominantly found on the face and neck.

The discoloration is caused by enlarged capillaries in the skin that result in increased blood flow. As capillary vessels are close to the surface of the skin, the increased blood flow gives the skin in affected areas a pink to purple appearance. Often a port wine stain is considered mostly cosmetic. However, over time the vessels can give a lumpy appearance to the skin. Also, if the stain covers an eye, or touches the nose or lip, this cobbling or thickening of the skin may impair vision and breathing in adulthood if the stain is left untreated.
Resources:
Cincinnati Children’s
Nova News
Vascular Birthmarks Foundation
Boston Children’s
Read blog posts about Port Wine Stain on No Hands But Ours.