Visit these blogs to read about families who have adopted children with this special need:
A Homespun Heritage
Dragons and Elephants
Happy Go Luckie
La Dolce Vita
Love You Forever
There’s No Place Like Home
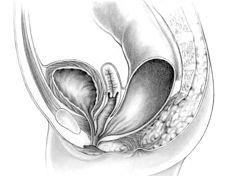
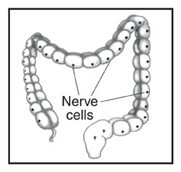
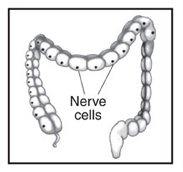
Anorectal Malformations (ARMs – sometimes referred to as anal atresia and imperforate anus) are defects that occur during the fifth to seventh weeks of fetal development. With these defects, the anus (opening at the end of the large intestine through which stool passes) and the rectum (area of the large intestine just above the anus) do not develop properly. Anorectal malformations affect 1 in 5,000 babies and is slightly more common in males. The exact cause of anorectal malformations is unknown.

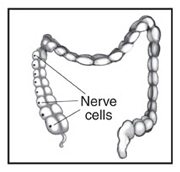
During a bowel movement, stool passes from the large intestine to the rectum and then to the anus. Nerves in the anal canal help us sense the need for a bowel movement and also stimulate muscle activity. Muscles in this area help control when we have a bowel movement.
With an anorectal malformation, any of the following abnormalities can occur:
– The anal passage may be narrow or misplaced in front of where it should be located
– A membrane may be present over the anal opening
– The rectum may not connect to the anus
– The rectum may connect to part of the urinary tract or the reproductive system through a passage called a fistula, and an anal opening is not present
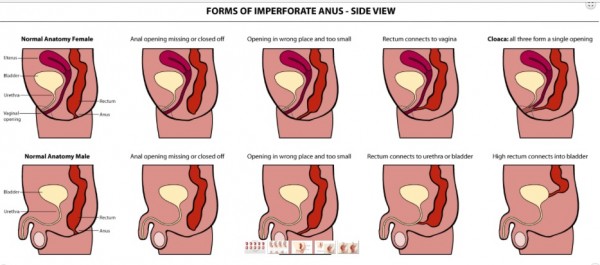
There are varying degrees of Anorectal Malformations (ARM) including high or low locations of the malformation. Children with low malformations are more prone to constipation (slow motility/hypomotile) while children with a shorter colon or a resection are more likely to have frequent, loose stooling (fast motility/hyermotile). A child born with an ARM defect will need surgery to correct the malformation and depending on how high the defect the child may need a temporary colostomy while the new anus heals. Some children from China come home with a colostomy and the remaining surgeries are done once home.
The surgery to correct an ARM is called the PSARP or posterior sagittal anorectalplasty. The PSARP approach was developed by Dr. Pena in 1980 and is also known as the Pena Pull-through. After the PSARP some children may require anal dilations to keep the new anus open.
While every ARM child’s condition is individual to them, many children can become “socially” continent through a bowel management program. A bowel management program can be tailored to each individual child and can include a daily stimulant laxative and/or daily enema. A few children are able to manage with diet alone but the majority of children will need a bowel management regimen.
When looking at the file of a child with an anorectal malformation it is important to remember that due to a lack of resources many ARM children are not properly maintained in China. These children may have had surgery, but present as incontinent. This could be due to surgery (which will require a re-do surgery once home), poor food choices (starches and other constipating foods), or lack of laxative/enema use. In addition, due to the chronic constipation found in many ARM children the child’s colon may be enlarged or stretched. ARM children’s colons will usually present as hypomotile (slow motility resulting in constipation) due to the original defect which affects the nerves and muscles. The stool moves very slowly eventually forming a stool ball making the child constipated. Over time, loose stool will begin leaking around the stool ball, and the child becomes incontinent due to the constipation. Children who have had their colons resected during a past surgery may present as hypermotile (fast motility resulting in frequent, loose stools). Many ARM children need either daily laxatives to help stimulate the colon to move, or daily enemas to fully empty the colon once a day. Once home, ARM children should be seen by a doctor well versed in ARMs to determine the best course of action. This plan includes having the child tested for VACTERL associated issues (tethered cord, heart, tracheoesophageal, renal/kidney and limb differences), a contrast enema, x-rays, and possibly a sedated exam to check the China repair and/or to better understand the child’s anatomy.
See also: Recto-vaginal fistula, Cloaca, Incontinence, and VACTERL.

ARM Expert Doctors, Centers, and Hospitals in the US:
Cincinnati Children’s Hospital Colorectal Center, Cincinnati, OH
Nationwide Children’s Hospital Center for Colorectal and Pelvic Reconstruction, Columbus, OH
Nemours/Alfred I. duPont Hospital for Children, Wilmington, DE – Dr. Charles Vinocur
Children’s Hospital Colorado, Aurora, CO
Seattle Children’s Hospital, Seattle, WA – Dr. Jeffrey Avansino
Morgan Stanley Children’s Hospital, New York NY – Dr. Shumyle Alam, urology specialist
Resources:
Yahoo! Group – IA Adoptive Parent Support
Yahoo! Group – IA Parental Support
Facebook Group – Imperforate Anus/Adoptive Mamas (Please note this is for adoptive moms only)
Imperforate Anus – Cincinnati Children’s
Imperforate Anus – Nationwide Children’s
Posterior Sagittal Anorectoplasty (PSARP)/Pull-through
Overview of ARMs
Bowel Management Information (Nationwide Children’s)
Dr. Dickie (Cincinnati Children’s) open Q & A webinar
Urological Concerns in Anorectal Malformations part 1, part 2
Gynecologic Concerns in Girls with ARMs
Article by Dr. Dickie (Cincinnati Children’s)
MACE (Malone)/Cecostomy
Additional Information about the Malone antegrade continence enema (MACE)
Children’s Hospital of Pittsburg
FAQs Bowel and Bladder Dysfunction adult adoptee, Alex Coe, answers FAQs
Pediatric Imperforate Anus Surgery
What Are Anorectal Malformations?
Bowel Management – an article by Drs. Levitt, Bischoff, & Pena
Pull-Thru Network
Continence Predictor Index
Read blog posts about Anorectal Malformations on No Hands But Ours.